di G. Bormetti, C.M. Carloni Calame, G. Montagna, N. Moreni, O. Nicrosini, M. Treccani
Esempi di complessità si trovano anche in biologia. Un caso interessante è l’alfabeto della vita. Cosa sappiamo?
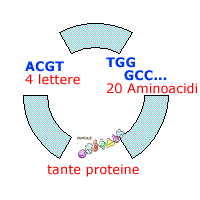
Le lettere fondamentali sono 4: A (Adenina), C (Citosina), G (Guanina) e T (Timina).
Il nostro DNA è una lunga sequenza di circa 3 miliardi di lettere, ma bisogna imparare a leggerle nel modo corretto.
Oggi sappiamo che le parole giuste sono quelle composte da tre lettere. Quante ne riuscite a costruire usando solo le lettere A C G e T? 43 = 64. Dobbiamo però eliminare dal calcolo 4 combinazioni che corrispondo alle sequenze che comunicano alla cellula di iniziare e terminare la sintesi delle proteine.
Le rimanenti 60 corrispondono solo a 20 aminoacidi ![]() differenti per cui è chiaro che triplette diverse tra loro possono avere lo stesso significato.
differenti per cui è chiaro che triplette diverse tra loro possono avere lo stesso significato.
Gli aminoacidi sono i mattoni fondamentali che costituiscono le proteine ![]() e che la cellula si preoccupa di assemblare nel modo corretto. Gli scienziati stanno cercando una procedura ottimale per prevedere le funzioni della proteina senza sintetizzarla ma semplicemente leggendo la sequenza di lettere corrispondente. Questo comporterebbe un notevole risparmio di tempo e di risorse. I problemi da affrontare sono tuttavia numerosi.
e che la cellula si preoccupa di assemblare nel modo corretto. Gli scienziati stanno cercando una procedura ottimale per prevedere le funzioni della proteina senza sintetizzarla ma semplicemente leggendo la sequenza di lettere corrispondente. Questo comporterebbe un notevole risparmio di tempo e di risorse. I problemi da affrontare sono tuttavia numerosi.
Solo il 5% del nostro DNA codifica per la sintesi delle proteine. Scoprire dove si nascondono le sequenze “utili” è un po’ come cercare un ago in un pagliaio e si rende necessario qualche stratagemma per semplificare la ricerca. Il problema di riconoscere delle strutture o evidenziare delle caratteristiche all’interno di un a enorme quantità di dati è un tema molto studiato. Nel nostro caso ci viene in aiuto una legge empirica scoperta da G.K. Zipf ![]() negli anni ’30 del secolo scorso.
negli anni ’30 del secolo scorso.
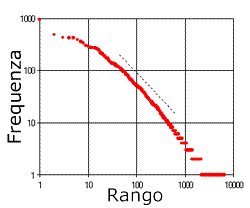
Provate a fare questo esperimento! Prendete un testo - un brano tratto da un romanzo di Dickens oppure da un testo scientifico - leggetelo e riscrivete a parte tutte le parole che vi compaiono. A fianco riportate il numero di volte che le avete incontrate. Naturalmente se il brano è molto lungo potete farvi aiutare in questo lavoro da un calcolatore. Ora mettetele in ordine, dalla più frequente alla più rara: la posizione in questa classifica definisce il rango r della parola. Se adesso riportate in ascissa il rango e in ordinata la frequenza f ottenete un grafico simile a quello riportato qui a fianco, che corrisponde alla legge:
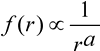
dove a è un numero positivo che prende il nome di esponente caratteristico.
Biologia e super-computer
Recentemente un gruppo di fisici ha scoperto che la stessa legge vale per le regioni di DNA non codificanti. Consideriamo adesso una sequenza AACGTTGCCGTGTAAACTGAC... e decidiamo, ad esempio, che le parole siano composte da 5 lettere. Adesso la nostra sequenza è un testo che possiamo analizzare come fatto nell’esperimento di G.K. Zipf. Con un computer e un po’ di pazienza siamo capaci di scoprire rapidamente dov’è la parte di DNA più interessante.
Se il grafico finale corrisponde alla legge di Zipf significa che abbiamo analizzato del DNA inutile per la sintesi delle proteine e possiamo scartare la sequenza. A questo punto osserviamo che questi esperimenti suggeriscono un’ipotesi molto affascinante. La maggior parte del DNA è non codificante ma non per questo necessariamente inutile. Essa condivide le stesse proprietà statistiche di un testo scritto da un essere umano e forse contiene delle informazionie che semplicemente non abbiamo ancora imparato a leggere.

Ora che sappiamo come individuare le regioni di DNA interessanti, si pone un secondo problema. Come possiamo capire quali saranno le proprietà delle proteine corrispondenti? I biologi hanno sviluppato delle tecniche molto raffinate per fare leggere agli apparati cellulari preposti le sequenze di basi. Una volta ottenuta la proteina, la sua struttura viene studiata con opportune tecniche cristallografiche per capire quali siano le sue funzioni. Questo procedimento è utile ma comporta dei tempi molto lunghi.
È importante, viste le ripercussioni in ambito medico e la possibilità di sviluppare nuove terapie, accelerare i tempi. La speranza per il futuro è quella che a fare tutte queste operazioni sia un calcolatore, che riceva come input la sequenza di basi e dia come risposta la struttura tridimensionale e le proprietà della proteina. Ma trovare le giuste combinazioni degli aminoacidi richiede grandi risorse di calcolo. A tale scopo sono stati sviluppati sia super-computer dedicati che strutture di calcolo distribuito su scala geografica.
Già oggi esistono super-computer che svolgono i numerosissimi calcoli necessari per comprendere come una proteina assuma una determinata configurazione tridimensionale, altamente complessa, come quelle mostrate nella figura accanto. Risolvere questo problema significherebbe capire come una proteina funziona, e questo aprirebbe nuove frontiere alla medicina e alla farmacologia.